|
|

|

|
| e-Pub |
Section: New Results
Refining the energy landscape sampling of protein-protein associations
Participants : Dmytro Kozakov, Leonard Jaillet.
PIPER is a FFT-based protein docking program with pairwise potentials. It combines a systematic sampling procedure with an original pairwise potential that provides an energy landscape representation through a set of samples [48].
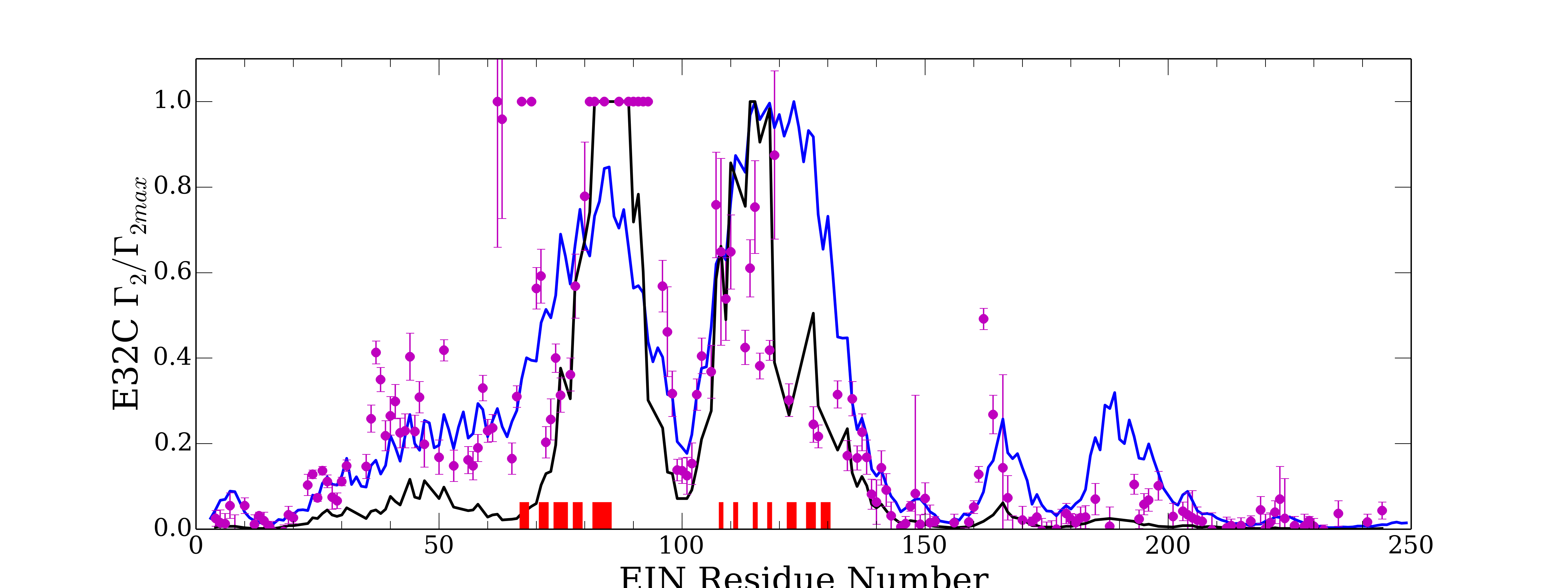
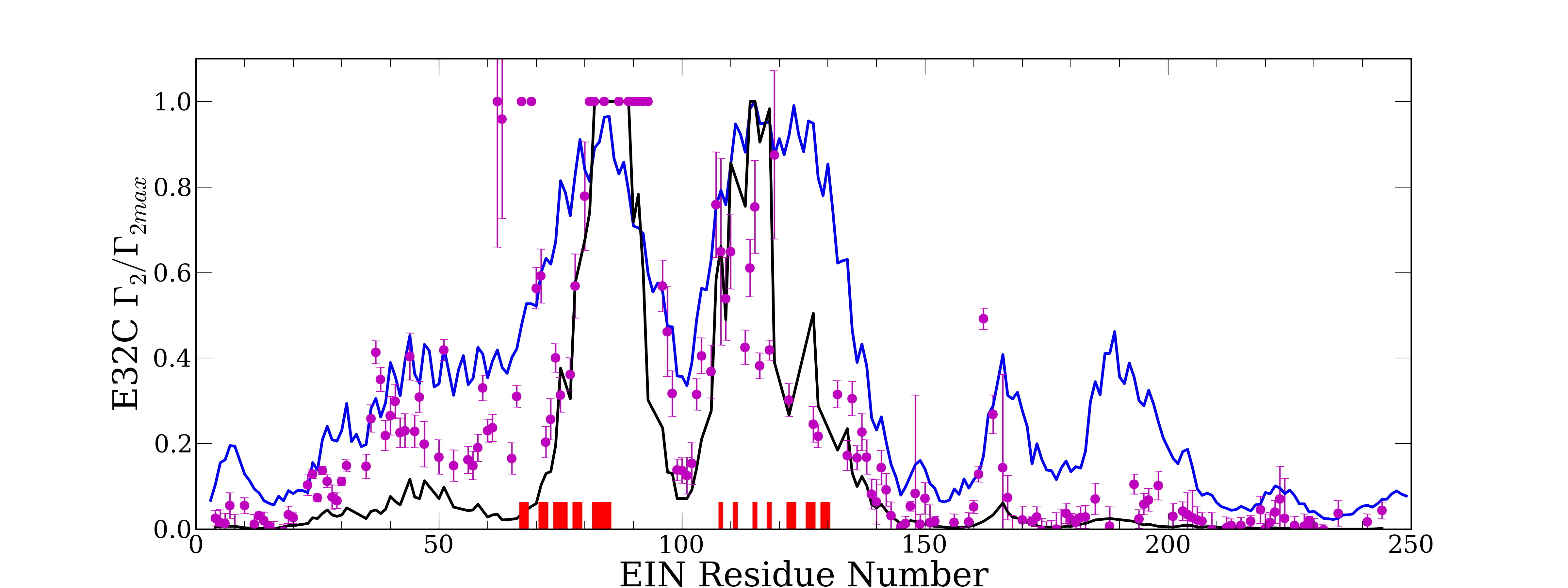
In [49], an experimental validation of the complexes obtained with PIPER, has been made possible thanks to the PRE method [31]. PRE (NMR paramagnetic relaxation enhancement) is an experimental technique used to characterize the states present for a given system. Hence, it characterizes the accessible region of the energy landscape corresponding to a given protein. For this, it introduces paramagnetic labels (tags) one at a time at few sites on one protein. The method then relies on measures of the transverse paramagnetic relaxation enhancement rates of the backbone amide protons (HN) of the partner protein. These value correspond to the weighted averages of the values for the various states present. One advantage of PRE is that it is nicely sensitive to lowly populated states.
In [49] the values measured obtained from a set of PIPER output have been compared to those obtained when using only the native state. It appears that using all the PIPER states give a better correlation respect to experimental results than when using only the native state.
In this context, our objective is to refine the energy landscape description by filtering some of the PIPER output complexes in order to improve even further the correlation with experimental measures. The method is developed as a module of the SAMSON software package (http://www.samson-connect.net/).
We have proposed a refinement from process of PIPER complexes based on two criterions: a RMSD-based filtering and an energy-based filtering.
The RMSD-filtering first creates a graph of connected component by connecting a pair of complexes if their distance is lower than a given RMSD threshold. Such a process forms clusters. Then, only the complexes that are in the cluster where belongs the native state are conserved. Since only rigid transforms are applied, RMSD are computed thanks to the fast RMSD computation method previously proposed in the team [56].
The energy-based filtering compares the energy of the complexes to the native state energy. The states for which the difference of energy is higher than a given threshold are discarded.
We have evaluated the results obtained when using our filtering scheme, for a distance threshold ranging form 3 to and for an energy threshold ranging from 70 to . Some setting of the filtering are able to improve the correlation (see figure 13), but the gain around remains limited (e.g. the correlation rising from to ). We are currently working on a more sophisticated state selection process to filter more precisely the PIPER states and hence to further improve the correlation.
|